
Giant Synovial Sarcoma of the Hand
Eugene F. Stautberg, MD¹; Sanjay Jain, MD²; Melissa Van Dellen, MD³
¹Department of Orthopaedic Surgery and Rehabilitation, The University of Texas Medical Branch; Galveston, TX
²Department of Radiology, The University of Texas Medical Branch; Galveston, TX
³Department of Pathology, The University of Texas Medical Branch; Galveston, TX
Corresponding Author:Eugene F. Stautberg, MD, Department of Orthopaedic Surgery and Rehabilitation, University of Texas Medical Branch, 301 University Blvd., Galveston, TX 77555-0165, USA; efstautb@utmb.edu
DOI: 10.18600/toj.010214
INTRODUCTION
Synovial sarcoma is a rare, malignant soft tissue sarcoma that, contrary to its name, does not originate from synovial cells. Synovial sarcoma typically occurs in males and females in the second through fourth decades of life and accounts for 5% to 10% of soft tissue sarcomas [1]. It is more common in the lower extremities, but it can occur around any joint. Patients typically present with a growing, painless mass, although larger masses abutting bones and nerves can cause pain. There can be a delay in diagnosis; one series of 5 patients showed the average duration of symptoms before diagnosis to be 8.2 months, with unplanned surgical excision in 2 of the patients [2]. Typically, synovial sarcomas are treated with wide surgical excision with possible radiotherapy and chemotherapy. We present a case of an initially indolent synovial sarcoma that transitioned into a rapidly growing mass, leading to an exceptionally large soft tissue burden. The patient had limited access to health care, leading to a delay in diagnosis and treatment, and the sarcoma recurred in the extremity despite an amputation with clear margins. CASE REPORT
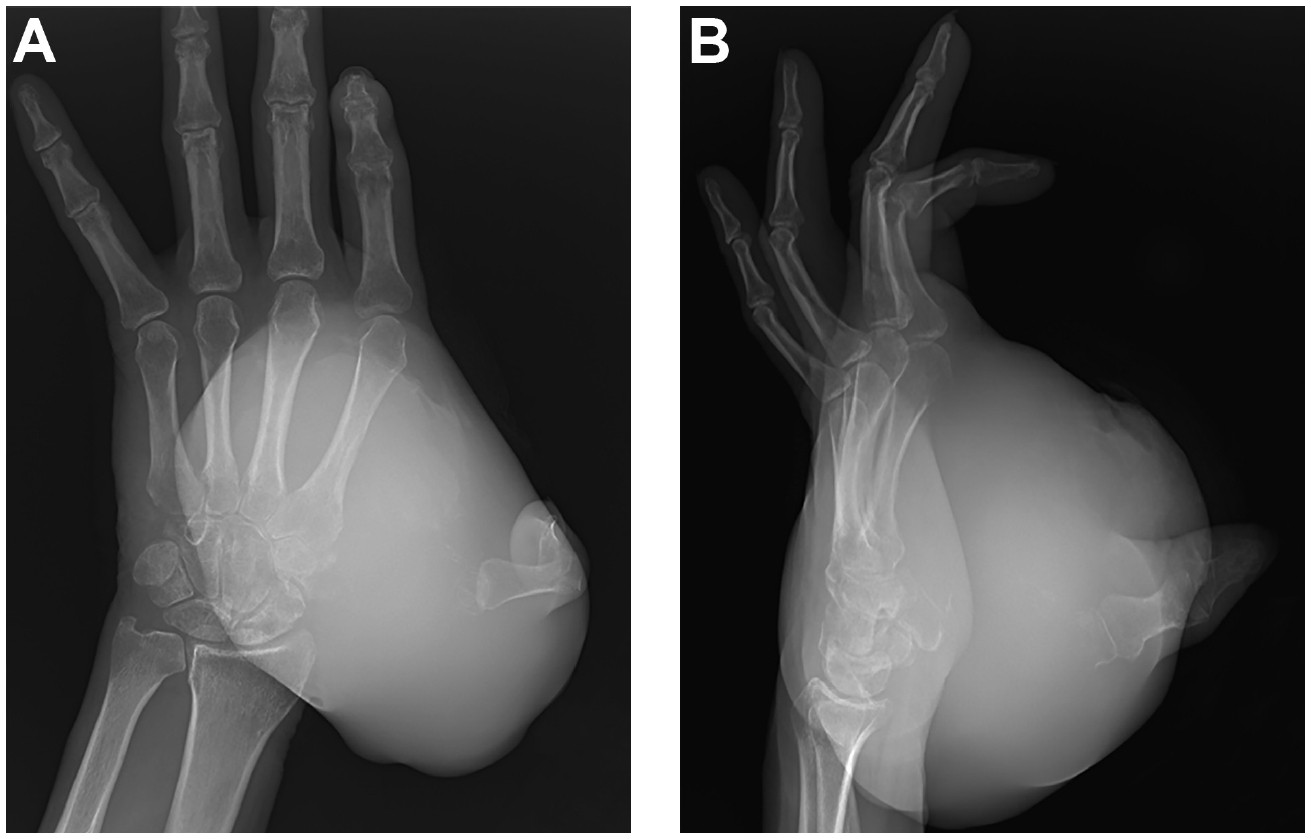
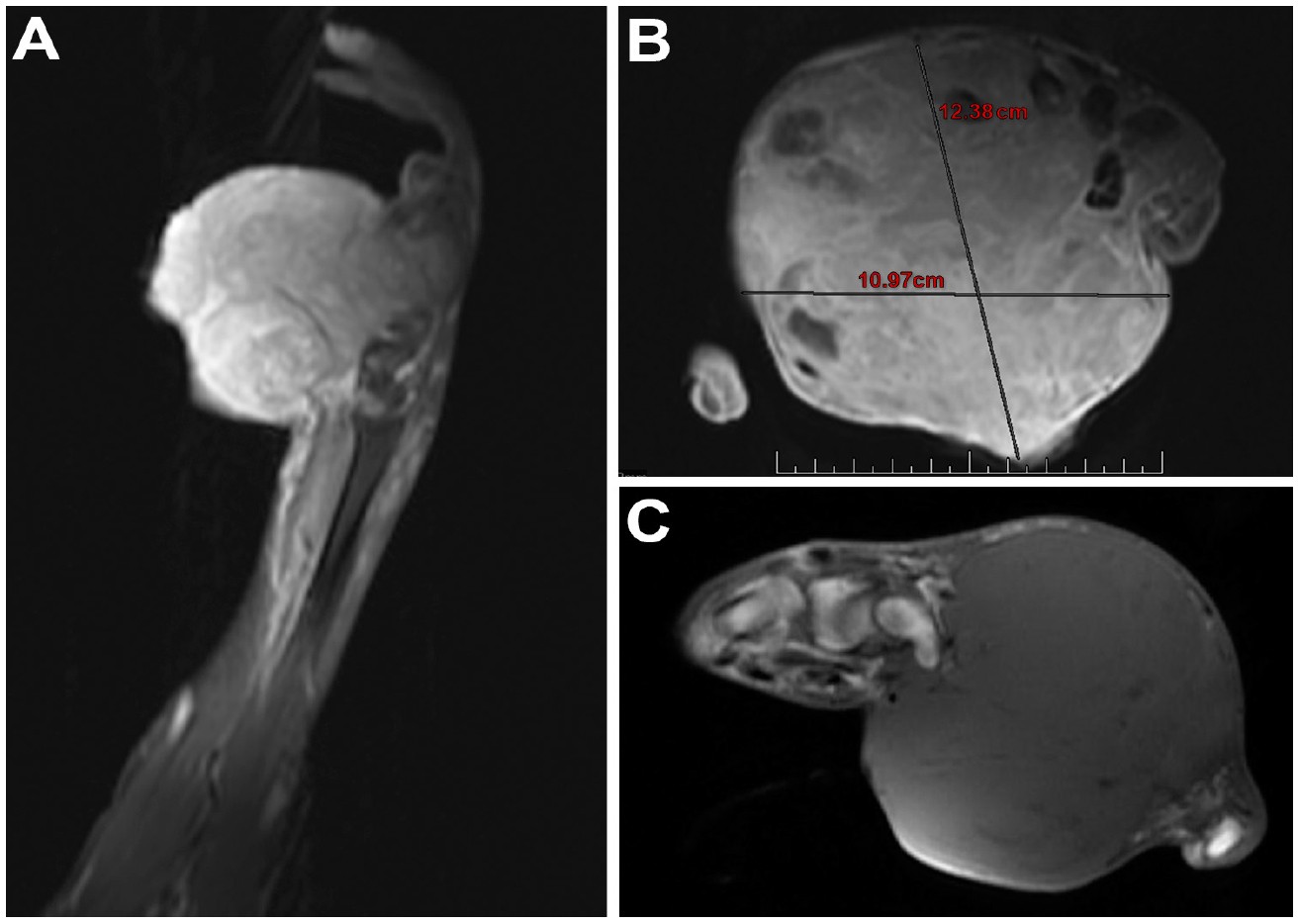
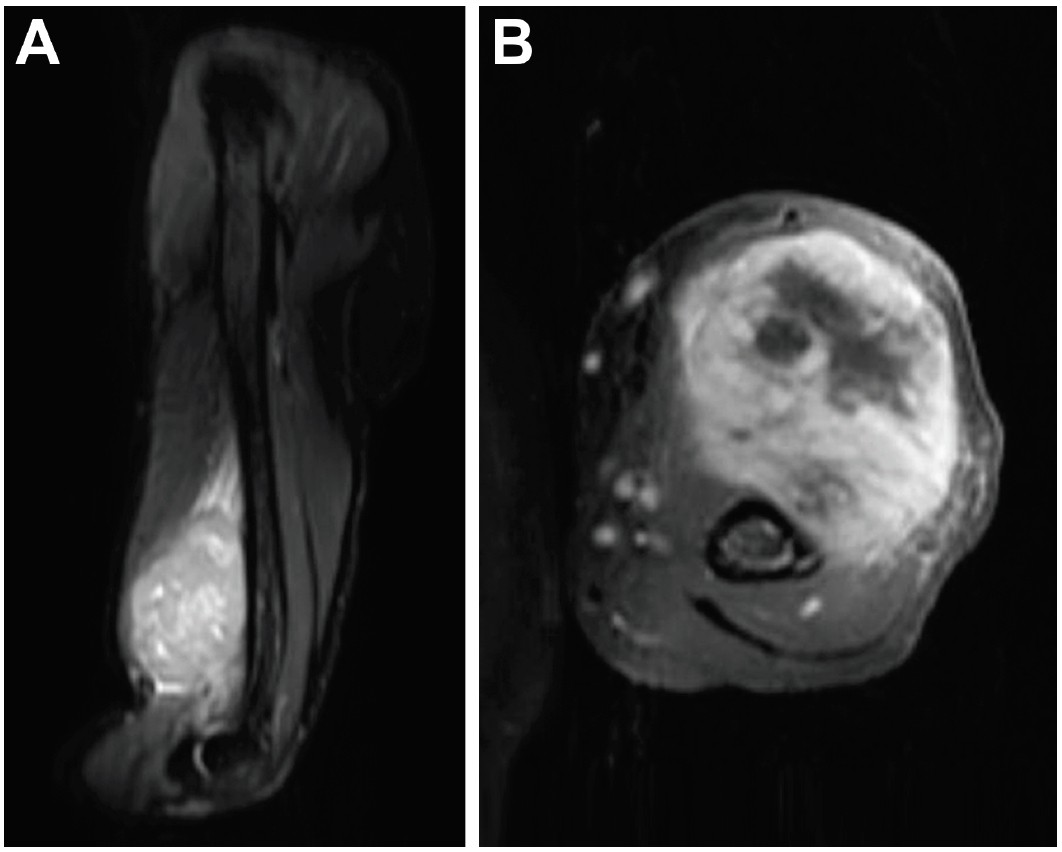
A 36-year-old male presented with a large left hand mass that had been present for 6 years. The patient was initially seen at an outside emergency department 1 year prior. The mass was centered over the volar, lateral aspect of the hand and was painless. At that time, a hematoma was diagnosed, and the mass was opened in the emergency department. Friable tissue was found, and pathology showed a malignant spindle cell monophasic neoplasm consistent with synovial sarcoma. However, the patient was uninsured at that time and did not follow up. Further, he became incarcerated and presented to our clinic 1 year after obtaining his diagnosis. At his initial presentation to our clinic, the mass had greatly increased in size since initial biopsy, leading to significant pain, thumb dislocation, and skin ulceration on the volar aspect of his hand. Plain radiographs showed a large mass of the left hand with associated osseous destruction and thumb dislocation (Figure 1). Magnetic resonance imaging (MRI) depicted a large, heterogeneously enhancing mass along the palmar surface of the left hand demonstrating heterogeneous T2 isointesity with areas of cystic formation (Figure 2). This mass measured 12x10x14 cm.
Synovial sarcoma is a rare, malignant soft tissue sarcoma that, contrary to its name, does not originate from synovial cells. Synovial sarcoma typically occurs in males and females in the second through fourth decades of life and accounts for 5% to 10% of soft tissue sarcomas [1]. It is more common in the lower extremities, but it can occur around any joint. Patients typically present with a growing, painless mass, although larger masses abutting bones and nerves can cause pain. There can be a delay in diagnosis; one series of 5 patients showed the average duration of symptoms before diagnosis to be 8.2 months, with unplanned surgical excision in 2 of the patients [2]. Typically, synovial sarcomas are treated with wide surgical excision with possible radiotherapy and chemotherapy. We present a case of an initially indolent synovial sarcoma that transitioned into a rapidly growing mass, leading to an exceptionally large soft tissue burden. The patient had limited access to health care, leading to a delay in diagnosis and treatment, and the sarcoma recurred in the extremity despite an amputation with clear margins. CASE REPORT
A 36-year-old male presented with a large left hand mass that had been present for 6 years. The patient was initially seen at an outside emergency department 1 year prior. The mass was centered over the volar, lateral aspect of the hand and was painless. At that time, a hematoma was diagnosed, and the mass was opened in the emergency department. Friable tissue was found, and pathology showed a malignant spindle cell monophasic neoplasm consistent with synovial sarcoma. However, the patient was uninsured at that time and did not follow up. Further, he became incarcerated and presented to our clinic 1 year after obtaining his diagnosis. At his initial presentation to our clinic, the mass had greatly increased in size since initial biopsy, leading to significant pain, thumb dislocation, and skin ulceration on the volar aspect of his hand. Plain radiographs showed a large mass of the left hand with associated osseous destruction and thumb dislocation (Figure 1). Magnetic resonance imaging (MRI) depicted a large, heterogeneously enhancing mass along the palmar surface of the left hand demonstrating heterogeneous T2 isointesity with areas of cystic formation (Figure 2). This mass measured 12x10x14 cm.
Computed tomography of the chest, abdomen, and pelvis did not show metastatic disease but did show enlarged left axillary lymph nodes. Lymph node core biopsy and fine-needle aspiration were performed to left axillary lymph nodes that did not show malignancy.
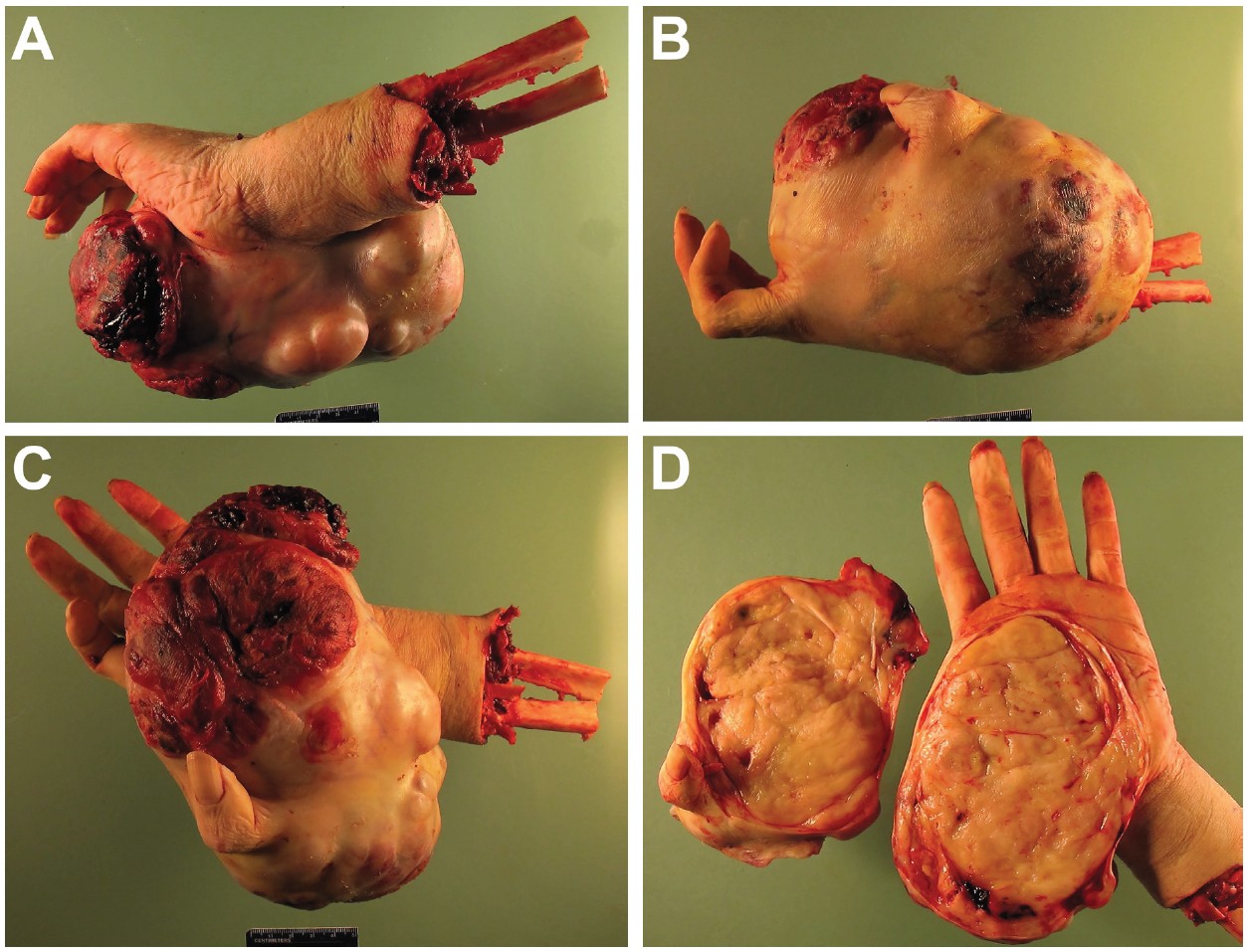
Further, over a 4-week period, pain became uncontrollable and the mass had begun to bleed from the ulcer. The patient required repeat blood transfusions. Because of the considerable size of the mass, continued blood loss, and recent drastic increase in the size of the mass, the patient was taken for transradial amputation (Figure 3) and preoperative chemotherapy was deferred. The patient tolerated this procedure well and recovery was uneventful.
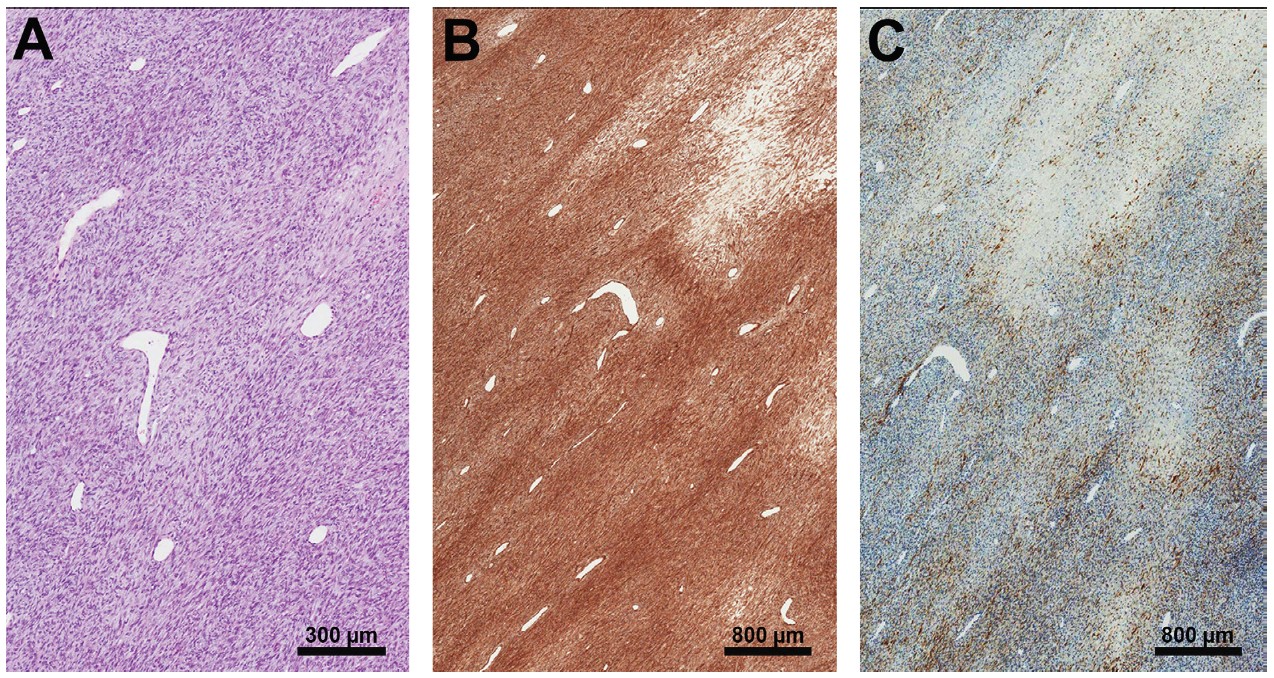
The gross pathology from the surgical specimen revealed a white-tan nodular lesion measuring 20 cm in greatest dimension with approximately 30% necrosis. Histology revealed intersecting fascicles of spindle cells, some with herringbone-type pattern, and round-oval vesicular nuclei. Occasional mast cells were identified, and mitoses were frequent (11 per 10 high-power fields). Immunohistochemistry showed the tumor cells to be diffusely and strongly positive for CD99 and focally positive for epithelial membrane antigen (EMA) (Figure 4). After transradial amputation, margins were noted to be negative, with 6 cm of clearance. FISH studies performed at the outside hospital on the preceding biopsy specimen revealed rearrangement for SS18 (SYT) in 81.5% of tumor cells, confirming the diagnosis of synovial sarcoma. There were no areas of epithelial component identified; therefore, the tumor was diagnosed as monophasic synovial sarcoma.
The patient was evaluated by general surgery for lymph node dissection, and based on negative imaging and biopsies, it was decided to defer lymph node dissection at that time. He was also evaluated by radiation oncology and medical oncology, and neither radiation therapy nor chemotherapy was recommended owing to wide surgical margins.
Further, over a 4-week period, pain became uncontrollable and the mass had begun to bleed from the ulcer. The patient required repeat blood transfusions. Because of the considerable size of the mass, continued blood loss, and recent drastic increase in the size of the mass, the patient was taken for transradial amputation (Figure 3) and preoperative chemotherapy was deferred. The patient tolerated this procedure well and recovery was uneventful.
The gross pathology from the surgical specimen revealed a white-tan nodular lesion measuring 20 cm in greatest dimension with approximately 30% necrosis. Histology revealed intersecting fascicles of spindle cells, some with herringbone-type pattern, and round-oval vesicular nuclei. Occasional mast cells were identified, and mitoses were frequent (11 per 10 high-power fields). Immunohistochemistry showed the tumor cells to be diffusely and strongly positive for CD99 and focally positive for epithelial membrane antigen (EMA) (Figure 4). After transradial amputation, margins were noted to be negative, with 6 cm of clearance. FISH studies performed at the outside hospital on the preceding biopsy specimen revealed rearrangement for SS18 (SYT) in 81.5% of tumor cells, confirming the diagnosis of synovial sarcoma. There were no areas of epithelial component identified; therefore, the tumor was diagnosed as monophasic synovial sarcoma.
The patient was evaluated by general surgery for lymph node dissection, and based on negative imaging and biopsies, it was decided to defer lymph node dissection at that time. He was also evaluated by radiation oncology and medical oncology, and neither radiation therapy nor chemotherapy was recommended owing to wide surgical margins.
Six months after transradial amputation, the patient presented with swelling and an irregular mass in the anterior, distal aspect of his arm (Figure 5). Core biopsy of that mass showed local recurrence of high-grade monophasic synovial sarcoma. Computed tomography of the chest showed consolidation of the lower lobe of the left lung, and the patient underwent bronchoscopy and transbronchial biopsy that showed infection without metastasis.
At this time, the patient had local recurrence without evidence of distant metastasis. Thus, surgical options were reviewed with the patient, including shoulder disarticulation versus forequarter amputation. The patient agreed to proceed with forequarter amputation as that would give the highest chance of complete tumor excision. Forequarter amputation was performed 9 months after initial amputation. Again, the patient’s postoperative course was uneventful. Radiation oncology and medical oncology re-evaluated the patient and did not recommend radiation therapy or chemotherapy. Currently, the patient has recovered from his forequarter amputation and is in routine surveillance for recurrence. DISCUSSION
Synovial sarcoma is a soft tissue neoplasm of mesenchymal stem cell origin [3]. Synovial sarcoma is a rare, malignant soft tissue sarcoma that typically presents in the distal portions of the extremities. Synovial sarcoma rarely originates inside of joints, but rather in proximity to joints. The name was given owing to its common location and histological resemblance to synovial cells. However, the name can be misleading, as this neoplasm is, in fact, not related to the synovium. Synovial sarcomas usually occur in the extremities, with the lower extremity accounting for approximately 60% to 70% of cases and the upper extremity accounting for 15% to 25% of cases.
At this time, the patient had local recurrence without evidence of distant metastasis. Thus, surgical options were reviewed with the patient, including shoulder disarticulation versus forequarter amputation. The patient agreed to proceed with forequarter amputation as that would give the highest chance of complete tumor excision. Forequarter amputation was performed 9 months after initial amputation. Again, the patient’s postoperative course was uneventful. Radiation oncology and medical oncology re-evaluated the patient and did not recommend radiation therapy or chemotherapy. Currently, the patient has recovered from his forequarter amputation and is in routine surveillance for recurrence. DISCUSSION
Synovial sarcoma is a soft tissue neoplasm of mesenchymal stem cell origin [3]. Synovial sarcoma is a rare, malignant soft tissue sarcoma that typically presents in the distal portions of the extremities. Synovial sarcoma rarely originates inside of joints, but rather in proximity to joints. The name was given owing to its common location and histological resemblance to synovial cells. However, the name can be misleading, as this neoplasm is, in fact, not related to the synovium. Synovial sarcomas usually occur in the extremities, with the lower extremity accounting for approximately 60% to 70% of cases and the upper extremity accounting for 15% to 25% of cases.
Synovial sarcoma can metastasize via lymph nodes and has a propensity to travel to the lungs [4]. Initial tumor size (>5 cm) and invasion of neurovascular structures are strong predictors of decreased survival [5,6]. In a review of 108 patients with local or locally recurrent synovial sarcoma without metastasis, the location and size of the tumor predicted outcomes. Among the 108 patients, survival rates at 5 and 10 years were 70% and 51%. If the mass was less than 5 cm, survival was 88% at 10 years, but for masses between 5 and 10 cm and those greater than 10 cm, the 10-year survival rates dropped to 38% and 8%. In an evaluation of the location of the lesion and prognosis, 10-year survival for distal tumors (hand and foot) was 65%, compared with 48% for tumors in the proximal extremities [7].
As with other soft tissue sarcomas, synovial sarcoma can start as a painless mass, leading to delay in diagnosis and treatment. Our case followed this pattern; however, once the mass began to cause discomfort, it grew rapidly, significantly increasing the deformity of the patient’s hand and destruction of soft tissues. Additionally, indolent masses in the extremities that slowly increase in size demand a high level of suspicion for malignancy to avoid inadvertent surgical violation of the mass, as happened in this case.
This case is unique as local failure after amputation is a rare complication. In a series of 44 synovial sarcomas, local control was obtained in all 8 treated with amputation [8]. Typically, local recurrences occur within a few years of limb salvage surgery; the same series showed a 72% local failure rate with wide local excision. Distant failure is more common than local recurrence.
As in other sarcomas, the lung is the most common site for distant metastasis. Care should be coordinated by a multidisciplinary team of medical oncologists, radiation oncologists, and oncologic surgeons. In our case, the surgical oncology team determined that a lymph node dissection was not indicated owing to a negative biopsy of lymph nodes. A review of sentinel lymph node biopsy (SLNB) in soft tissue sarcomas without known metastasis showed positive sentinel nodes in 2 of 42 patients with synovial sarcoma [9]. Regional lymph node metastasis developed in 1 patient with negative SLNB. It was concluded that SLNB is not an important diagnostic tool in synovial sarcoma [9]. Further, because of wide surgical margins in both cases, medical oncology and radiation oncology determined that no further treatment was needed. Radiographs can appear normal in approximately 50% of cases, particularly with small lesions. In 10% to 25% of cases, radiographs can show changes that include pressure erosions or periosteal reaction. Calcifications are seen in about one third of cases. Extensive calcifications within a lesion may be associated with an improved prognosis [10]. Magnetic resonance imaging is the optimal modality to evaluate the anatomic extent and intrinsic characteristics of the lesion, including any neurovascular encasement. Synovial sarcoma will typically demonstrate heterogeneously T2 hyperintesity compared with skeletal muscle. The mass will demonstrate enhancement and appear highly complex, possibly containing 3 different signal intensities, termed the “triple” sign [11]. This finding is due to the combination of solid tumor, hemorrhage, and necrosis. Hemosiderin, cystic change, and fluid-fluid levels are common. T1-weighted images may demonstrate a “split fat” sign, which is a thin rim of fat around the mass due to the intermuscular origin near a neurovascular bundle.
As in our case, the monophasic fibrous type is essentially devoid of the epithelial component, and consists of intersecting fascicles of fibrous cells, sometimes with a focal whorling pattern. A herringbone-type pattern reminiscent of fibrosarcoma or nuclear palisading resembling a neural pattern is not uncommon. Additionally, perivascular hyalinization, hemangiopericytoma-like vessels, and mast cell infiltrate may be seen [12]. Biphasic synovial sarcoma consists of fascicles of spindle cells as well as groups of epithelial cells. The epithelial cells are cuboidal to columnar, with oval nuclei, and may be arranged in nests, cords, tubules, or papillae. Eosinophilic secretions associated with the epithelial component are not uncommon. Monophasic synovial sarcoma can be further categorized into fibrous type and epithelial type, the latter being the most rare presentation.
Immunohistochemically, synovial sarcomas are typically reactive for epithelial membrane antigen (EMA), cytokeratin, and CD99. Some tumors show focal positivity for S100.
Characteristic cytogenetic and molecular findings show a balanced reciprocal translocation t(X;18)(p11.2;q11.2), involving the SYT gene on chromosome 18, and either the SSX1 or SSX2 gene on the X chromosome. When subtypes of synovial sarcoma were reviewed, a SYT-SSX1 fusion transcript was detected in 64% of synovial sarcomas, all biphasic; a SYT-SSX2 fusion transcript was detected in 36% of tumors, all monophasic [13]. Patients with a SYT-SSX2 transcript had significantly better metastasis-free survival than those with a SYT-SSX1 transcript [14]. In summary, this case shows a giant synovial sarcoma that remained indolent for a number of years before turning aggressive and rapidly growing. A high index of suspicion must be given to slow-growing, painless masses, particularly if over 5 cm, in a young population to avoid inadvertent biopsy and delay in diagnosis. Also, this case showed an instance of local failure after amputation, despite negative margins. Further, providers must recognize when a patient with an aggressive disease has limited access to health care, and they must work with the patient to overcome these barriers. REFERENCES
[1] Kransdorf MJ. Malignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and location. AJR Am J Roentgenol 1995;164(1):129-34.
[2] Outani H, Hamada K, Oshima K, Joyama S, Naka N, Araki N, Ueda T, Yoshikawa H. Clinical outcomes for patients with synovial sarcoma of the hand. Springerplus. 2014;3:649.
[3] Naka N, Takenaka S, Araki N, Miwa T, Hashimoto N, Yoshioka K, Joyama S, Hamada K, Tsukamoto Y, Tomita Y, Ueda T, Yoshikawa H, Itoh K. Synovial sarcoma is a stem cell malignancy. Stem Cells. 2010;28(7):1119-31.
[4] Bakri A, Shinagare AB, Krajewski KM, Howard SA, Jagannathan JP, Hornick JL, Ramaiya NH. Synovial sarcoma: imaging features of common and uncommon primary sites, metastatic patterns, and treatment response. AJR Am J Roentgenol. 2012;199(2):W208-15.
[5] Lewis JJ, Antonescu CR, Leung DH, Blumberg D, Healey JH, Woodruff JM, Brennan MF. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol. 2000;18(10):2087-94.
[6] Thompson RC, Garg A, Goswitz J, Cheng EY, Clohisy DR, Dusenbery K. Synovial sarcoma. Large size predicts poor outcome. Clin Orthop Relat Res. 2000;373:18-24.
[7] Deshmkh R, Mankin HJ, Singer S. Synovial sarcoma: the importance of size and location for survival. Clin Orthop Relat Res. 2004;(419):155-61.
[8] Paulino AC. Synovial sarcoma prognostic factors and patterns of failure. Am J Clin Oncol. 2004;27(2):122-7.
[9] Andreou D, Boldt H, Werner M, Hamann C, Pink D, Tunn PU. Sentinel node biopsy in soft tissue sarcoma subtypes with a high propensity for regional lymphatic spread–results of a large prospective trial. Ann Oncol. 2013;24(5):1400-5.
[10] Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. From the archives of the AFIP: Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-65.
[11] Dhawan A, Shenoy AM, Chavan P, Sandhu S, Sriprakash D.Synovial sarcoma of the infratemporal fossa with extension into the oral cavity—a rare presentation and literature review. J Oral Maxillofac Surg. 2012;70(12):2923-9.
[12] Goldblum JR, Weiss SW, Folpe AL. Enzinger and Weiss’s Soft Tissue Tumors, 6th ed. Philadelphia, PA, Saunders/Elsevier. 2014.
[13] Törnkvist M, Brodin B, Bartolazzi A, Larsson O. A novel type of SYT/SSX fusion: methodological and biological implications. Mod Pathol. 2002;15(6):679-85.
[14] Kawai A, Woodruff J, Healey JH, Brennan MF, Antonescu CR, Ladanyi M. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998;338(3):153-60.
As with other soft tissue sarcomas, synovial sarcoma can start as a painless mass, leading to delay in diagnosis and treatment. Our case followed this pattern; however, once the mass began to cause discomfort, it grew rapidly, significantly increasing the deformity of the patient’s hand and destruction of soft tissues. Additionally, indolent masses in the extremities that slowly increase in size demand a high level of suspicion for malignancy to avoid inadvertent surgical violation of the mass, as happened in this case.
This case is unique as local failure after amputation is a rare complication. In a series of 44 synovial sarcomas, local control was obtained in all 8 treated with amputation [8]. Typically, local recurrences occur within a few years of limb salvage surgery; the same series showed a 72% local failure rate with wide local excision. Distant failure is more common than local recurrence.
As in other sarcomas, the lung is the most common site for distant metastasis. Care should be coordinated by a multidisciplinary team of medical oncologists, radiation oncologists, and oncologic surgeons. In our case, the surgical oncology team determined that a lymph node dissection was not indicated owing to a negative biopsy of lymph nodes. A review of sentinel lymph node biopsy (SLNB) in soft tissue sarcomas without known metastasis showed positive sentinel nodes in 2 of 42 patients with synovial sarcoma [9]. Regional lymph node metastasis developed in 1 patient with negative SLNB. It was concluded that SLNB is not an important diagnostic tool in synovial sarcoma [9]. Further, because of wide surgical margins in both cases, medical oncology and radiation oncology determined that no further treatment was needed. Radiographs can appear normal in approximately 50% of cases, particularly with small lesions. In 10% to 25% of cases, radiographs can show changes that include pressure erosions or periosteal reaction. Calcifications are seen in about one third of cases. Extensive calcifications within a lesion may be associated with an improved prognosis [10]. Magnetic resonance imaging is the optimal modality to evaluate the anatomic extent and intrinsic characteristics of the lesion, including any neurovascular encasement. Synovial sarcoma will typically demonstrate heterogeneously T2 hyperintesity compared with skeletal muscle. The mass will demonstrate enhancement and appear highly complex, possibly containing 3 different signal intensities, termed the “triple” sign [11]. This finding is due to the combination of solid tumor, hemorrhage, and necrosis. Hemosiderin, cystic change, and fluid-fluid levels are common. T1-weighted images may demonstrate a “split fat” sign, which is a thin rim of fat around the mass due to the intermuscular origin near a neurovascular bundle.
As in our case, the monophasic fibrous type is essentially devoid of the epithelial component, and consists of intersecting fascicles of fibrous cells, sometimes with a focal whorling pattern. A herringbone-type pattern reminiscent of fibrosarcoma or nuclear palisading resembling a neural pattern is not uncommon. Additionally, perivascular hyalinization, hemangiopericytoma-like vessels, and mast cell infiltrate may be seen [12]. Biphasic synovial sarcoma consists of fascicles of spindle cells as well as groups of epithelial cells. The epithelial cells are cuboidal to columnar, with oval nuclei, and may be arranged in nests, cords, tubules, or papillae. Eosinophilic secretions associated with the epithelial component are not uncommon. Monophasic synovial sarcoma can be further categorized into fibrous type and epithelial type, the latter being the most rare presentation.
Immunohistochemically, synovial sarcomas are typically reactive for epithelial membrane antigen (EMA), cytokeratin, and CD99. Some tumors show focal positivity for S100.
Characteristic cytogenetic and molecular findings show a balanced reciprocal translocation t(X;18)(p11.2;q11.2), involving the SYT gene on chromosome 18, and either the SSX1 or SSX2 gene on the X chromosome. When subtypes of synovial sarcoma were reviewed, a SYT-SSX1 fusion transcript was detected in 64% of synovial sarcomas, all biphasic; a SYT-SSX2 fusion transcript was detected in 36% of tumors, all monophasic [13]. Patients with a SYT-SSX2 transcript had significantly better metastasis-free survival than those with a SYT-SSX1 transcript [14]. In summary, this case shows a giant synovial sarcoma that remained indolent for a number of years before turning aggressive and rapidly growing. A high index of suspicion must be given to slow-growing, painless masses, particularly if over 5 cm, in a young population to avoid inadvertent biopsy and delay in diagnosis. Also, this case showed an instance of local failure after amputation, despite negative margins. Further, providers must recognize when a patient with an aggressive disease has limited access to health care, and they must work with the patient to overcome these barriers. REFERENCES
[1] Kransdorf MJ. Malignant soft-tissue tumors in a large referral population: distribution of diagnoses by age, sex, and location. AJR Am J Roentgenol 1995;164(1):129-34.
[2] Outani H, Hamada K, Oshima K, Joyama S, Naka N, Araki N, Ueda T, Yoshikawa H. Clinical outcomes for patients with synovial sarcoma of the hand. Springerplus. 2014;3:649.
[3] Naka N, Takenaka S, Araki N, Miwa T, Hashimoto N, Yoshioka K, Joyama S, Hamada K, Tsukamoto Y, Tomita Y, Ueda T, Yoshikawa H, Itoh K. Synovial sarcoma is a stem cell malignancy. Stem Cells. 2010;28(7):1119-31.
[4] Bakri A, Shinagare AB, Krajewski KM, Howard SA, Jagannathan JP, Hornick JL, Ramaiya NH. Synovial sarcoma: imaging features of common and uncommon primary sites, metastatic patterns, and treatment response. AJR Am J Roentgenol. 2012;199(2):W208-15.
[5] Lewis JJ, Antonescu CR, Leung DH, Blumberg D, Healey JH, Woodruff JM, Brennan MF. Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol. 2000;18(10):2087-94.
[6] Thompson RC, Garg A, Goswitz J, Cheng EY, Clohisy DR, Dusenbery K. Synovial sarcoma. Large size predicts poor outcome. Clin Orthop Relat Res. 2000;373:18-24.
[7] Deshmkh R, Mankin HJ, Singer S. Synovial sarcoma: the importance of size and location for survival. Clin Orthop Relat Res. 2004;(419):155-61.
[8] Paulino AC. Synovial sarcoma prognostic factors and patterns of failure. Am J Clin Oncol. 2004;27(2):122-7.
[9] Andreou D, Boldt H, Werner M, Hamann C, Pink D, Tunn PU. Sentinel node biopsy in soft tissue sarcoma subtypes with a high propensity for regional lymphatic spread–results of a large prospective trial. Ann Oncol. 2013;24(5):1400-5.
[10] Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. From the archives of the AFIP: Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-65.
[11] Dhawan A, Shenoy AM, Chavan P, Sandhu S, Sriprakash D.Synovial sarcoma of the infratemporal fossa with extension into the oral cavity—a rare presentation and literature review. J Oral Maxillofac Surg. 2012;70(12):2923-9.
[12] Goldblum JR, Weiss SW, Folpe AL. Enzinger and Weiss’s Soft Tissue Tumors, 6th ed. Philadelphia, PA, Saunders/Elsevier. 2014.
[13] Törnkvist M, Brodin B, Bartolazzi A, Larsson O. A novel type of SYT/SSX fusion: methodological and biological implications. Mod Pathol. 2002;15(6):679-85.
[14] Kawai A, Woodruff J, Healey JH, Brennan MF, Antonescu CR, Ladanyi M. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med. 1998;338(3):153-60.